Selectivity and molecular mechanism of the Au(III)-catalyzed [3+2] cycloaddition reaction between (<i>Z</i>)-<i>C</i>,<i>N</i>-diphenylnitrone and nitroethene in the light of the molecular electron density theory computational study
Keywords:
Au(III) complexes, nitroalkene, [3+2] cycloaddition, mechanism, molecular electron density theoryAbstract
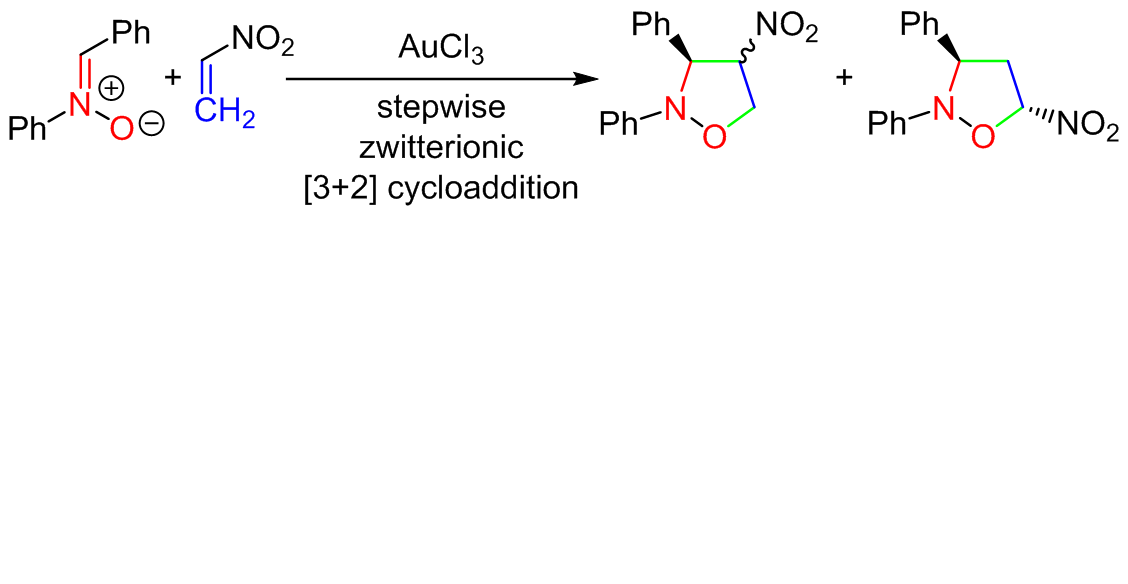
The regio- and stereoselectivity and the molecular mechanism of the Au(III)-catalyzed [3+2] cycloaddition reaction between (Z)C,Ndiphenylnitrone and nitroethene in the benzene solution were evaluated on the basis of the B3LYP18-D3(BJ)/def2svp (PCM) quantum-chemical calculations. It was found, that the presence of the Au(III) molecular segments in the reaction medium changes dramatically the cycloaddition mechanism. In particular, the observed single-step mechanism under the thermal conditions is replaced to stepwise, zwitterionic mechanism in three from four theoretically possible paths.
Downloads
Published
2025-02-13
