COMPUTATIONAL STRUCTURE – BIOLOGICAL ACTIVITY AND RETROSYNTHESIS INVESTIGATIONS OF 1,2,3-TRIAZOLE-QUINOLINE HYBRID MOLECULES AS POTENTIAL RESPIRATORY VIRUS INHIBITORS
Keywords:
triazole-quinoline hybrid molecules, ADMET, molecular docking, molecular dynamics, retrosynthesis, SARS-CoV-2Abstract
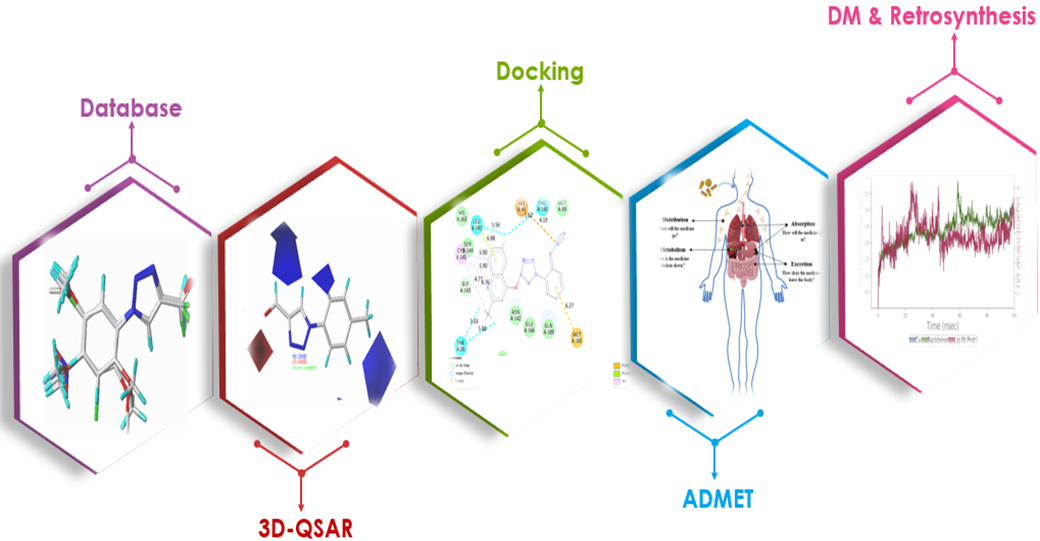
SARS-CoV-2 infection is a severe public health problem that motivates researchers to find new antiviral drugs for effective therapy. A computational study was carried out for disubstituted 1,2,3-triazole-quinoline hybrid molecules as SARS-CoV-2 inhibitors using the 3D-QSAR approach to investigate the quantitative structure – biological activity relationship with regard to Mpro protease inhibition. Whithin the 3D-QSAR approach, Comparative Molecular Field Analysis (CoMFA) and Comparative Molecular Similarity Index Analysis (CoMSIA) were performed, where CoMFA model values are Q2 0.53, R2 0.97 and the best values of the CoMSIA model are Q2 0.67, R2 0.93). Molecular docking and molecular dynamics analysis were carried out to study the stability of the ligands inside the Mpro active site, and an in silico ADMET study explored the pharmacological and physicochemical properties of the proposed molecules as new antiviral agents. Subsequently, the CoMFA and CoMSIA models were evaluated by an external validation using the A. Golbraikh and A. Tropsha criteria. A molecular docking study was carried out for 18 derivatives of 1,2,3-triazole-quinoline and 5 of them were proposed as new Mpro biological target candidates. Molecular dynamics calculations permitted to assess the reliability of this proposition, and the ADMET study evaluated the physicochemical properties and drug similarity of the proposed structures. Finally, the two online platforms were used to suggest the best possible synthesis route based on the retrosynthetic analysis.