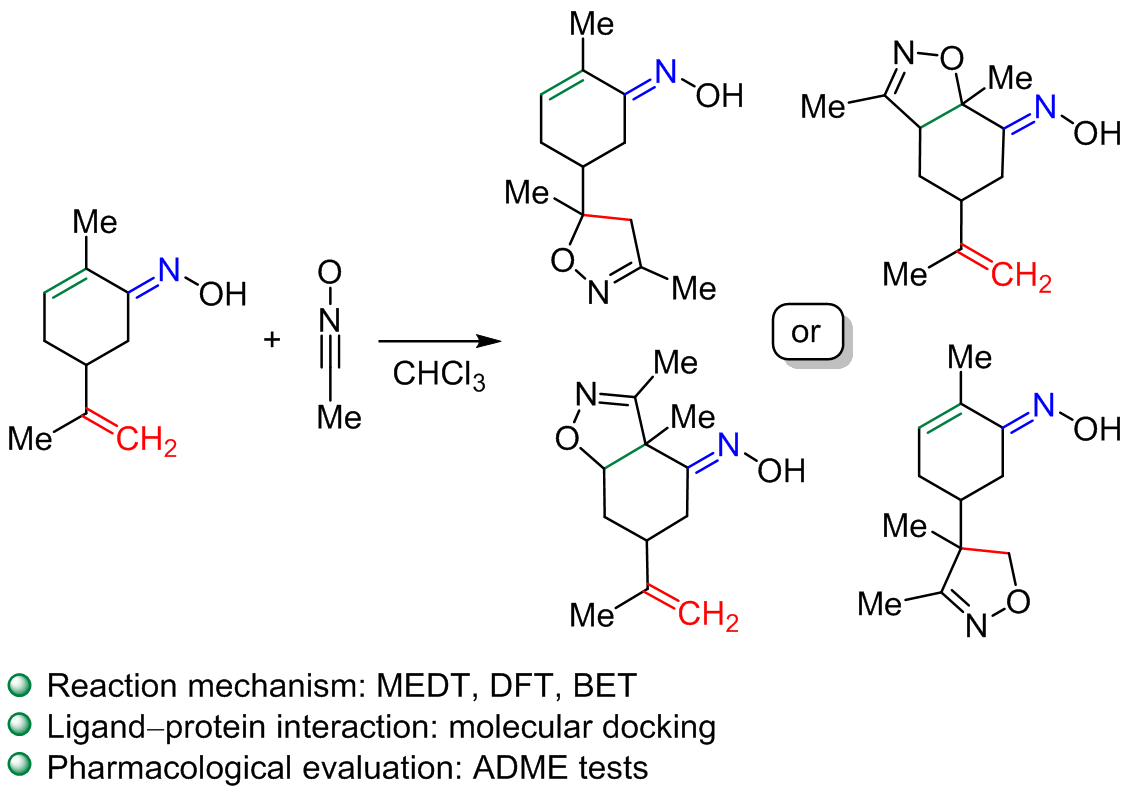
Quantum study of the [3+2] cycloaddition of nitrile oxide and carvone oxime: insights into toxicity, pharmacokinetics, and mechanism
Ключевые слова:
carvone, nitrile oxide, oxime, ELF, MEDT, molecular docking, regiospecificityАннотация
The study employs molecular electron density theory (MEDT) to investigate the mechanism of [3+2] cycloaddition involving carvone-based oxime and nitrile oxide. Reactivity indices as well as activation and reaction energies were determined by density functional theory (DFT) calculations employing the B3LYP/6-311(d,p) method. According to a conceptual DFT indices study, carvone-based oxime was characterized as a nucleophile, while the investigated nitrile oxide acts as an electrophile in the reaction. The reaction demonstrates chemoselectivity and regiospecificity, as confirmed by the electron localization analysis and energy evaluations, consistent with experimental observations. Bond evolution theory analysis suggests a one-step two-stage mechanism with asynchronous bond formation. Additionally, docking studies on the possible reaction products reveal enhanced interactions with proteins, attributed to the presence of oxygen and nitrogen atoms, which increase their affinity for the SARS-CoV-2 protease. Furthermore, a docking analysis shows that one of the investigated ligands exhibits strong binding affinity for both HIV-1 and SARS-CoV-2 proteins, indicating a high potential for interaction with these viral targets.